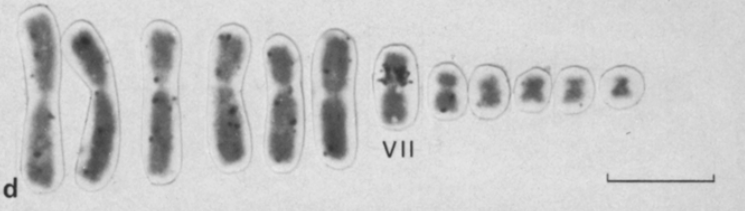
The information reported in the two previous posts is useful to take two important steps in the de-novo genome sequencing. First, in the computational demanding assembling step, the lower the intraspecific variability is, the easier it will be to pile up raw reads and recognize stretches of contiguous sequences. Therefore, a specimen with low genetic variability (low number of differences between maternal and paternal chromosomes) should be used. We will therefore use a an individual from the Northern population. Second, the size of the genome and the low number of chromosomes make this species a good candidate to use an approach called “single chromosome sequencing”, that consists in physically isolate each chromosome through microdissection and sequence each chromosome separately. This is a relatively new approach used to target single chromosome with the aim of building a reference genome for species with very large genomes. The technique was successfully used for instance in Amphibia in 2015 by Keinath and colleagues, that isolated and sequenced two chromosomes from the Mexican axolotl. The isolation procedure will be carried out using a micromanipulator connected to a microscope. The microdissected chromosome will be placed into a microtube to be used for downstream steps. These include the amplification of the selected genomic material, usually through a Whole Genome Amplification kit, the evaluation of the amount of total DNA obtained and the final sequencing of the genomic subset.
As you might be wondering, the recognition of the different chromosomes is done on sight, but fortunately some previous studies showed that B. pachypus chromosomes are different enough from each other, in terms of size and shape, and therefore will be relatively easy to isolate.
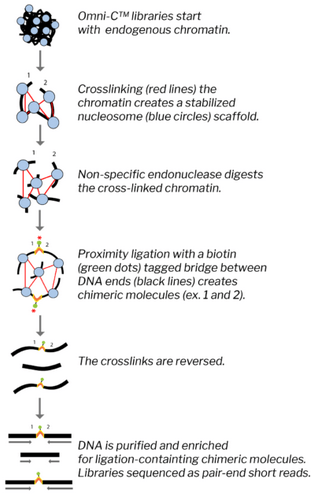
The amplified DNA will be then fragmented and read with the Illumina technology, producing a dataset of pair end reads, each 250 bp long. We aim at obtaining an average coverage of 25X for each chromosome library. While reducing the genome complexity through single chromosome sequencing ease dramatically the process of genome characterization, the sizes of the single chromosomes are still quite big and require other specific approaches to help the assembly. For this reason, and in particular to be further aware of the connections between genetic region, another recently developed approach will be implemented, called Omni-C. This technique allows to understand the relative proximity of genetic regions within a scaffold. Briefly, the protocol starts with chromatin extracted from cell cultures, blood or tissue. The nucleosome is then fixed with crosslinks that, even after the nucleases digest the crosslinked chromatin, fragments are kept in place (i.e. fragments close to each other in the scaffold will remain so despite being “free”). The free ends are then labelled with biotin and ligated. In this step the probability that two fragments are ligated is proportional to how close to each other they are in the nucleosome. Only fragment that originate from the ligatin process are then amplified and analyzed. As for the previous library, also Omni-C library will be sequenced with Illumina pair end chemistry, aiming at 25X coverage.
References
– Keinath, M. C., Timoshevskiy, V. A., Timoshevskaya, N. Y., Tsonis, P. A., Voss, S. R., & Smith, J. J. (2015). Initial characterization of the large genome of the salamander Ambystoma mexicanum using shotgun and laser capture chromosome sequencing. Scientific reports, 5, 16413
– Vitelli, L., Batistoni, R., Andronico, F., Nardi, I., & Barsacchi-Pilone, G. (1982). Chromosomal localization of 18S+ 28S and 5S ribosomal RNA genes in evolutionarily diverse anuran amphibians. Chromosoma, 84(4), 475-491.
