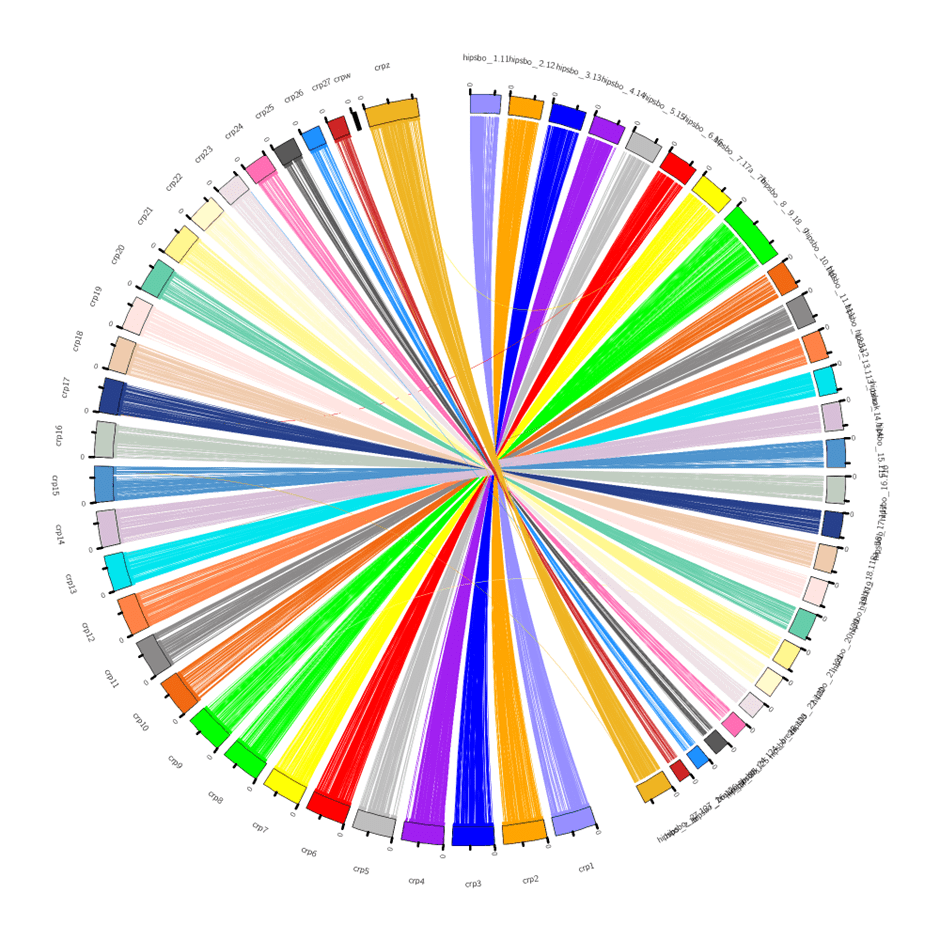
We are finalizing the genome assembly of the Ponza grayling Hipparchia sbordonii, which is currently undergoing a manual refinement step. A comparative assessed of different de novo assembly strategies identified Canu as the best-performing algorithm: following different polishing and scaffolding steps, the genome assembly included 99 scaffolds (N50 = 10019938, L50 = 28 and BUSCO completeness = 98.6%). This was definitely a good achievement, even though the quality of the assembly still did not reach a chromosome level!
Thus, we decided to exploit synteny data to improve the contiguity of the Ponza grayling assembly, using the available high-quality genomes of Pararge aegeria (which belongs to the same subfamily Satyrinae) as a reference to build a map of orthologous genes. The Canu assembly was improved by using the scaffolds generated with other assembly approaches for gap-filling, whenever possible. This manual curation step brought the assembly to a near-chromosome level, significantly improving all relevant metrics (N50 = 14947868, L50 = 11 and a BUSCO = 98.7%). The recent publication of the genome of the congeneric species H. semele will allow us to further validate the reliability of this approach.